Stargardt's Disease
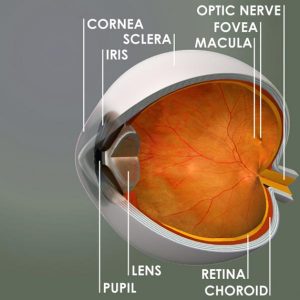
1 in 10,000 people has Stargardt’s disease. It is an inherited disease that is sometimes, but not always, characterized by the distribution of yellow “flecks” throughout the fundus, or back wall, of the eye and a rapidly progressive loss of central vision.
Inherited eye conditions usually present themselves early in life. The symptoms of Stargardt’s disease typically occur between seven and 12 years of age. When Stargardt’s disease develops in adulthood, it is sometimes referred to instead as fundus flavimaculatus (FFM), which is a subtype of Stargardt’s disease. In most cases of Stargardt’s and FFM, yellow deposits of a protein called lipofuscein are scattered throughout the macula. The subsequent scarring and deterioriation of the macula result in central vision loss that may progress quickly or over many years. Half of those with Stargardt’s disease are legally blind by the age of 50, but some peripheral vision remains intact. There is no standard treatment for Stargardt’s disease, but low-vision aids can help an individual to retain the ability to perform some work-related and daily activities.
Some forms of Stargardt’s disease are inherited from a parent who has the disease, but Stargardt’s disease can also appear in the child of parents who have normal retinas and no vision problems. This happens because the defective gene that causes Stargardt’s disease is recessive; it will not be physically expressed unless the other of the pair is also recessive. A parent with normal vision can carry the gene for Stargardt’s disease, but it is inactive. When two parents carry the inactive gene, there is a one in four chance that the two recessive genes will pair-up, and Stargardt’s disease will be expressed.
Genetic counseling can be very helpful in family planning, both for parents of a child with Stargardt’s disease who are considering having more children, and for an adult with Stargardt’s disease who would like to start a family.